Fuel & fuel cycle
Actinide recycling within closed fuel cycles
14 February 2012The global energy context argues in favour of the sustainable development of nuclear energy, since the demand for energy will significantly increase, while resources will tend to get scarcer. Reprocessing and recycling nuclear fuel, together with fast reactors, can help nuclear power to conserve existing uranium resources and reduce the nuclear waste burden for future generations. By Christophe Poinssot and Bernard Boullis
Several kinds of nuclear fuel cycles are implemented today: most countries chose the so-called once-through cycle which basically considers spent nuclear fuel as waste, whereas others like France, UK, Japan and soon China reprocess their spent fuel to recover the energetically-valuable material Pu (and partially U) to produce Mixed Oxide Fuel (MOX) to be irradiated in a second cycle (a twice-through cycle). None of them allow a complete use of the natural resource; when discharged from reactor, 96% of spent nuclear fuel is still composed of U and Pu which can produce electricity and could be recycled.
France chose Pu mono-recycling in LWR reactors more than 20 years ago and implements it with the Areva La Hague reprocessing plant and the Marcoule MELOX recycling plant, which both have run with success for more than 15 years. More than 26,000 tHM of UOX spent fuels have been treated in the UP2 and UP3 plants (La Hague), and the corresponding Pu inventory has been recycled as MOX fuels that are produced in the MELOX plant. MOX fuel is burnt in 22 French pressurized water reactors (but over 40 reactors worldwide use MOX fuel). More than 6000 MOX fuel assemblies have been produced at the MELOX plant, for instance.
Treatment and recycling requires (i) dissolving the spent nuclear fuel to access the nuclear materials and (ii) extracting the valuable elements, U and Pu, from the ultimate waste, mainly fission products and minor actinides. The PUREX process is currently used to separate the U and Pu. This process uses an organic solvent (a mixture of tributyl phosphate (TBP) and diluent), that selectively extracts the uranium and plutonium from other fission products, which remain in the aqueous phase. The process is carried out in counter-current devices such as pulsed columns or mixer-settlers in order to increase the effectiveness of separation. In the La Hague plants for instance, both recovery yields and purification levels are very high (up to 99.9% of uranium and plutonium recovered and decontamination factors about 106 or more). A final conversion step allows production of oxide powders which are compatible with the fabrication specification of the fuel fabrication process, thanks to the precipitation of intermediate oxalates.
The amount of secondary waste, the level of radioactivity released and workers’ radiation exposure have been drastically lowered during the last two decades, although in the same period throughput was significantly increased. Such a fuel cycle saves up to 17% of the natural uranium consumed in a once-through cycle; indeed, in MOX fuels, the natural U-235 fissile isotope is replaced by recycled Pu-239 and depleted uranium is used as a matrix. In France for instance, roughly 1500t of natural uranium is saved annually thanks to Pu mono-recycling and MOX fabrication.
Recycling drastically decreases not only the volume, but also the long-term radiotoxicity of the final waste. Indeed, the long-term toxicity of spent nuclear fuel is dominated by plutonium (Figure 2). Recycling plutonium leads to a long-term radiotoxicity decrease of one order of magnitude; that is, it will need 10 times less to return to the radiotoxicity of the initial natural uranium. Also, it provides an effective burning of fissile plutonium and avoids its accumulation in spent fuel stockpiles, decreasing the risk of diversion.
Finally, Pu mono-recycling frees the ultimate waste (fission products and minor actinides) from the residual properties of the spent nuclear fuel. They are to be conditioned in a borosilicate nuclear glass, in France the so-called R7T7, whose lifetime has been demonstrated to be in the range of 106 years in French future repository conditions. Furthermore, such confinement material does not have any instant release fraction (IRF), whereas the IRF significantly contributes to the long-term impact of spent nuclear fuel.
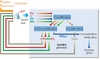
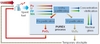
The COEXTMprocess
In the near future, improvement for Pu mono-recycling will be related to the implementation of the COEXTM process, which allows a complete co-management of U and Pu within the fuel cycle (Figure 3). First, it will produce homogeneous (U,Pu)O2 powders and subsequent fuel pellets, instead of PuO2 and subsequent heterogeneous pellets. Second, it prevents the existence of any pure Pu stockpile in the fuel cycle.
The PUREX extraction scheme has been modified to allow part of the uranium to follow the Pu flux in the partitioning step. Its efficiency has been demonstrated on actual spent fuel dissolution solutions in the Atalante hot laboratories in CEA Marcoule.
In terms of the conversion step, R&D performed in the last decade has demonstrated that the current oxalic conversion process can be adapted to the production of (U,Pu)O2 thanks to the existence of perfect (U,Pu) solid solutions both for the oxalates UIV2-xPuIIIx(M+)2+x(C2O4)5,nH2O [1, 2] and the oxides (U,Pu)O2 [3]. The conversion can therefore be realized by the precipitation of (UIV,PuIII)oxalate which are thereafter calcinated to oxides at 700-800°C. Basic R&D focused on the determination of the crystallographic structures of the different end-members and solid solutions existing between PuIII-UIV and the quantification of the different kinetics steps (especially nucleation and growth). In particular, it has shown a pseudomorphic transformation in which the morphology of the final oxide powders is defined by those of the oxalate powders. Similar developments have been performed on the calcination steps. All these results allow the derivation of a global mechanistic understanding of the oxalic UPu co-conversion. It is currently under implementation in a predictive modeling tool, and will optimize the process chemical conditions as a function of the properties of the oxide powders.
Significant amounts of co-converted powders have been produced in the Atalante hot laboratory, so the powder can be demonstrated to meet the requirements of the MELOX fabrication process and is suitable for subsequent MOX fuel fabrication [4]. Furthermore, experimental pellets have been produced and irradiated in the CEA Phénix sodium fast reactor in 2008 (COPIX experiments). Post-irradiation examinations are planned in the next few years.
As a conclusion, the COEXTM process is now ready for implementation at the industrial scale. The remaining R&D is only related to optimization of technological devices.
Fast reactors
Although U-238 represents 99.2% of natural uranium, it is not fissile. It could be fertilised by neutron capture in order to produce Pu-239 which is fissile, and work with an implementation of Pu multi-recycling. This is however not possible in LWRs since neutron capture of U-238 is not efficient enough and the neutron capture of uneven isotopes of plutonium is high, leading to the formation of minor actinides. On the other hand, fast neutron spectra relatively increase the capture of neutrons by U-238, leading to the formation of plutonium isotopes which are all fissile in such conditions. For example, the ratio of the capture to fission cross sections of Pu-238, Pu-240 and Pu-242 are increased in fast spectra compared to thermal spectra by a factor of 22, 250 and 36 respectively. In conclusion, fast neutron spectra allow the effective consumption of U-238 to produce fissile plutonium isotopes which are subsequently fissioned to produce energy and electricity. Reactors using fast neutrons are hence potentially able to use more than 80% of the natural resources instead of < 1% for LWR.
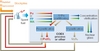
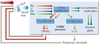
However, implementation of fast neutron reactors (FR) is intrinsically linked to the implementation of reprocessing and recycling facilities. Indeed, such nuclear systems require recycling the Pu fissile elements from spent fuel into freshly-fabricated fuel. On the other hand, the front end of the fuel cycle can be significantly reduced since no additional uranium ore is theoretically necessary; indeed, stockpiles of depleted uranium and reprocessed uranium can be used and represent a very significant reserve, up to several thousand years in France, for instance.
For the fuel cycle, the main evolution required deals with completely recovering Pu and U from MOX fuels. For the separation processes, the same process as in the previous step, COEXTM, can be implemented (Figure 4). Feasibility is therefore already achieved. Treatment of FR MOX fuels and subsequent Pu multi-recycling has already been demonstrated at the industrial scale in France. Indeed, 27t of FR spent MOX fuels have already been reprocessed in the APM (Marcoule) and UP2-400 (La Hague) plants in the 1980s and 1990s.
Beyond the numerous technical issues related to the development of SFRs, some scientific and technological issues are still to be addressed for the fuel cycle. On a first approximation, they are mainly related to the front end of the process and not to extraction. Indeed, both the higher Pu content in FR fuels and the larger mass of metallic materials in FR fuels will impact the spent fuel mechanical shearing and dissolution processes.
First, to access the fuel pellets and the nuclear materials, structural materials have to be removed. They can represent up to 70% of the total spent fuel mass (~30% in the case of LWR spent fuels) and are composed of stainless steel, which is more sensitive to corrosion in highly concentrated nitric acid than zircaloy is in LWR fuels. The technological process therefore needs to be optimized to allow access to the oxide fuels while minimising the amount of metallic material sent to dissolution. (In a previous industrial campaign, mechanical removal of hexagonal tubes was implemented). Furthermore, the cladding material for FR fuel is still under study. Among others, oxide dispersed steels (ODS) are serious candidates since they are more resistant to higher irradiation. However, their resistance to corrosion in nitric acid still needs to be quantified [5].
Second, the dissolution kinetics of (U,Pu)O2 is strongly dependent on the Pu content, and strongly decreases for Pu content higher than 35%. Due to the higher Pu content of FR MOX fuels, dissolution can locally be kinetically inhibited locally by a high Pu content. Additional basic R&D on the actual mechanisms governing the dissolution of (U,Pu)O2 oxides and irradiated fuels still needs to be performed.
Third, when dissolution is achieved, solutions can be highly concentrated in cations that can re-precipitate. This phenomenon has already been observed during the reprocessing of LWR spent fuels by the PUREX process. Such secondary phases are important to identify and to quantify since they can incorporate some nuclear materials, modify the global nuclear material mass balance, and therefore affect the criticality management and the efficiency of the recovery process. Here again, basic studies are necessary to derive the thermodynamic and kinetic properties of the relevant secondary phases to be considered in such conditions.
As a second priority, R&D is still necessary to adapt the extraction scheme to the peculiarities of the FR MOX fuel, in particular its high Pu content, and the potential presence of metallic elements coming from partial dissolution of fuel cladding. At this stage, any potential innovation for enhancing the global process efficiency and separation process ease of implementation are of interest, since they can help the economics of recycling and nuclear energy. Basic R&D as well as process design are under study.
Recycling minor actinides
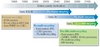
Recycling minor actinides would dramatically reduce the residual toxicity and the subsequent lifetime of the final waste; some hundreds of years would be sufficient to decrease the residual radiotoxicity down to that of the natural uranium ore (Figure 5). Decreasing the waste heat power by recycling minor actinides, particularly americium, would allow a denser repository and significantly increase the waste volume to be stored in the same repository by a factor up to eight.
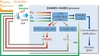
A wide-ranging research programme has been executed in France since 1991 in the framework of two successive waste management acts [6]. Recycling minor actinides involves first recovering them from the spent nuclear fuel after dissolution, and then recycling and burning them in nuclear reactors. There are two main options considered in current research: heterogeneous and homogeneous recycling [7], which differ in the way the material is put in the reactor core.
Each process would require a different separation process. All of the separation processes are based on hydrometallurgical processes familiar to operating reprocessing plants. Furthermore, they allow quite high separation yields (>106) and they produce a low amount of secondary waste. However, they require extensive R&D to define and select the most appropriate extraction molecules and to design the associated process. This R&D is based on a sophisticated partitioning chemistry under highly radioactive conditions. A three-step methodology is usually applied: (i) explorative R&D on more than 100 potential molecules to gain an in-depth understanding of the actual molecular mechanisms, (ii) batch lab experiments in active conditions to derive relevant parameters to design the extracting processes and (iii) demonstration experiments on spent nuclear fuels at the kilogram scale. Most of this effort was shared at the European level through several EU-funded projects, as EUROPART, PARTNEW and the current ACSEPT project.
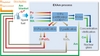
Heterogeneous recycling aims to recover minor actinides Am and Cm in a dedicated flux and recycle them in specific target or blanket with a relatively high content (up to 20%) at the periphery of the reactor core (Figure 6).
Minor actinide recycling has been investigated at the CEA for the two last decades [8]. The technological feasibility of Am and Cm recovery was demonstrated in 2005 through the treatment of 13 kg of genuine spent nuclear fuel in the Atalante facility with high yields of actinide recovery (>99.9%). To accomplish this, two processes were successively implemented: first, DIAMEX to coextract trivalent 4f and 5f elements (respectively Ln(III) and An(III)), and second, SANEX, to selectively strip An(III) with a polyaminocarboxylate compound used in a buffered medium around pH = 3 (with a citrate buffer for example). In order to develop a simpler single-cycle An (III) partitioning process, new extracting molecules have been investigated at CEA. The TetraOctyl-DiGlycolAmide (TODGA) molecule, which presents a very high extraction affinity towards trivalent actinides and lanthanides, appears very promising [9]. A simplified flowsheet has been designed and consists of three main steps [10]:
1. Trivalent actinides and lanthanides are coextracted by TODGA/TBP solvent
2. A hydrophilic polyaminocarboxylate complexing agent (diethylenetriamine-pentaacetic acid or DTPA) selectively strips the An(III) in a buffered solution (malonic acid at pH 2), while the Ln(III) remains complexed by TODGA in the organic solvent thanks to a salting-out agent in the aqueous phase.
3. Finally, diluted nitric acid strips the lanthanides.
A hot test using a genuine PUREX raffinate was performed in 2009 in the Atalante facility. This counter-current hot test was succesful; recovery yields of An(III) larger than 99.9% were obtained with high decontamination factors of Ln(III) (less than 5% mass of Ln in the An product) [11].
A specific option of heterogeneous recycling would be to recycle solely Am, since its contribution to the residual heat power and radiotoxicity is higher than Cm, and it is quite a bit easier to manage than Cm. This would simplify fuel manufacturing while allowing a subsequent potential benefit for a repository.
The principle of the EXAm process is based on the extraction of americium together with some light lanthanides having close values of distribution coefficients in high nitric acidity, while curium and other lanthanides remain in the aqueous phase (Figure 7). The TODGA amide molecule is added in the aqueous phase, in order to increase the selectivities of Am/Cm and Am/heavy lanthanides, because of the preferential complexation of curium and heavy lanthanides by this diglycol-amide; its addition largely improves the global efficiency of the process, and in doing so decreases the number of separation stages. Then Am is selectively stripped from light lanthanides as in GANEX step two (see below).
Homogeneous recycling
Homogeneous recycling aims to recover all the actinides, Pu and Am and Cm, in a single flux, and recycle them at a low concentration (up to 2%) in all the fuel in the reactor core. Grouped actinides can be separated through the so-called GANEX process (Group ActiNide EXtraction) which aims to recover all of the transuranium elements from the high-activity spent fuel solution. It consists of two steps: selective extraction of uranium, and then partitioning of actinides from fission products and lanthanides (Figure 8).
The selective separation of uranium(VI) is performed by a hydrometallurgical process using a monoamide extractant DEHiBA (N,N-di-(ethyl-2-hexyl)isobutyramide) diluted in an industrial aliphatic solvent HTP. Based on batch experimental results, a physicochemical model was developed to describe the extraction of U, Np, Pu, and Tc by DEHiBA. With the help of this model, a flowsheet was designed and tested in 28 mixer-settlers (laboratory scale) on a genuine high-liquid waste in the CBP hot cell (Atalante facility) in June 2008. After running 60 hours, more than 99.99% of the initial uranium was recovered with a good purity; there was a very low residual content of transuranium elements and fission products [12].
The GANEX step two process consists of a grouped separation of transuranium elements (Np, Pu, Am, and Cm) from the fission products by solvent extraction. It was derived from the DIAMEX-SANEX process by adjusting experimental conditions (in particular, selection of complexing agents and optimization of reagent concentrations) [13]. In terms of results, Np, Pu, Am and Cm were recovered together in a single flux (actinide product) and the losses of transuranium in the different outputs and in the solvent were estimated at a value lower than 0.5% (essentially neptunium) at the end of the test, corresponding to a recovery yield of actinides higher than 99.5%.
After 20 years of research, France and other countries have developed some relevant and effective processes to selectively partition minor actinides in view of their subsequent recycling through fission in reactors. Technical feasibility can therefore be considered as achieved. However, several technological and scientific issues are still under investigation to obtain efficient and relevant processes at the industrial scale, in particular with regard to the understanding of the actual molecular processes and origin of selectivity, chemical process simulation, solvent cleanup and continuous industrial contactors (for example, pulsed column, mixer-settlers, and so on).
Transmutation
Neutronics and fuel R&D has been supported in many countries for more than 20 years to demonstrate the feasibility of minor actinide transmutation. A massive research programme on transmutation fuels and targets, including in particular an experimental irradiation programme, has been carried out since the 1990s on both a national and international level (in particular within EU projects). This programme was based on irradiation tests in the European HFR in Petten and in Phénix from 2003 to 2009. In particular, flux conditions of Phénix were well-suited to study of irradiation damage of fuels and targets for transmutation under representative conditions of fast- and partly-moderated flux, which are considered to be the most efficient for transmutation of MA and some long-lived fission products. More than 35 irradiation experiments were conducted for both homogeneous and heterogeneous transmutation during that period, some of which are still under post-irradiation characterization.
In the case of heterogeneous transmutation, minor actinides are not located within the core of the reactor, but at its periphery. Therefore they do not interact much with the neutronic behaviour of the core, but rather use external neutrons coming out of the core. In this case, the main limitation is related both to the behaviour of the matrix containing the MA and the global balance between the available neutrons and those that would be consumed by the MA for transmutation. Two options are under consideration: either an inert matrix which is used for a target, or fertile UO2 which is used as a blanket. The main issue to address is the production of a large amount of He due to the high MA content, and its behaviour within the fuel rod.
In the case of homogeneous transmutation, minor actinides are aimed to be dispersed in the whole core at a constant loading. Therefore, the main restriction to introducing minor actinides into critical reactors is linked to their impact on the core’s reactivity and kinetic parameters. They would tend to produce a drop in the fuel temperature coefficients, an increase in reactivity linked to coolant voiding, and a reduction of delayed neutron yield. Therefore, the MA content has to be limited to around 2-3% of total heavy nuclides for the reference large sodium-cooled reactors.
Conclusion
Three main improvement steps in nuclear fuel reprocessing and recycling, including a gradual transformation to fast reactors starting in the 2020s, can be defined for the next century (Figure 9). Current once-through fuel cycles without any recycling misuse the uranium natural resource with an efficiency that is lower than 1%. This practice clearly does not preserve the rights of future generations to use nuclear fission to produce electricity. Furthermore, it yields a massive accumulation of spent nuclear fuel which is transferred as a burden towards future generations, instead of recycling the valuable materials and confining as best as possible the ultimate waste in a specifically-tailored wasteform such as nuclear glass. In the near future, the increase in energy demand and increased need to mitigate climate change should yield development of a partially-closed fuel cycle that includes recycling strategies and Generation IV fast reactors.
Author Info:
Christophe Poinssot, CEA Radiochemistry and Processes Department head; visiting professor of actinide materials chemistry, University of Sheffield, UK Bernard Boullis, programme director, fuel cycle technologies and waste management, CEA. Both authors are also professors at the National Institute for Nuclear Science and Technology, France.
This article is based on a paper presented at ICAPP 2011 which took place on 2-5 May 2011 in Nice, France.
This article was originally published in the January 2012 issue of Nuclear Engineering International magazine.
References
[1] S. GRANDJEAN, B. ARAB-CHAPELET, G. LETURCQ, S. PICART, P. BARON, P. BLANC, M. MASSON, "Synthesis of Mixed Uranium Plutonium Oxide by Oxalic Co-conversion as Precursors for Advanced MOX fabrication, International Conference ATALANTE 2008: Nuclear Fuel Cycles for a Sustainable Future, May 19-23, 2008, Montpellier (France), (2008).
[2] S. GRANDJEAN, B. ARAB-CHAPELET, A.C. ROBISSON, F. ABRAHAM, PH. MARTIN, J-PH. DANCAUSSE, N. HERLET, C. LÃORIER, "Structure of mixed U(IV)-An(III) precursors synthesized by co-conversion methods (where An=Pu, Am or Cm)", Journal of Nuclear Materials 385, 204-207, (2009).
[3] P.M. MARTIN, S. GRANDJEAN, B. ARAB-CHAPELET, A-C ROBISSON, G. LETURCQ, A.C. SCHEINHOST, A. ROSSBERG, "Homogeneity at molecular scale of (U,Pu)O2 solid solutions probed by XAS", Actinides 2009, San Francisco, California, USA, July 12-17, (2009).
[4] R. CASTELLI , T GERVAIS, D FAVET, B YTOURNEL, COEXTM Pellet Fabrications In MELOX Test Chain, Proceedings of GLOBAL 2009, Paris, France, Sept. 6-11, (2009).
[5] M.AUROY, B. GWINNER, D. MAS, F. MISERQUE, M. TABARANT, On the corrosion behavior of martensitic/ferritic steels in nitric media, Oral communication in the 2nd international conference "Corrosion and Material Protection", 19-22th April 2010, Prague, (2010).
[6] D.WARIN, Status of the French research program on P&T, Journal of Nuclear Science and Technology, 44(3), Pages 410-414, (2007).
[7] C.POINSSOT, D.WARIN, C.ROSTAING, "Recent achievements towards the recycling of minor actinides for the improvement of future nuclear fuel cycle", European Nuclear Conference ENC 2010 transactions, June 2010, Barcelona, (2010).
[8] C. ROSTAING, P. BARON, B. LORRAIN, D. WARIN, B. BOULLIS, "Minor actinides partitioning : main results during fiftenn years research and prospects", European Nuclear Conference (ENC - 2007), 16/09/07 au 20/09/07, Bruxelles, Belgique (2007).
[9] Y SASAKI, Y SUGO, H SUZUKI, T, KIMURA, "Development of ARTIST process, extraction and separation of actinides and fission products by TODGA", Conference ATALANTE 2004, Names, Gard (France), 21-25 juin 2004, (2004).
[10] C. HILL, M. MIGUIRDITICHIAN, X. HÃRÈS, C. SOREL, I. BISEL, D. ESPINOUX, P. BARON, "A new concept for an(iii)/ln(iii) separation using todga extractant", 10th Information Exchange Meeting on Actinide and Fission Product Partitioning and Transmutation â€" 2008, Mito, Japon (2008).
[11] C. HILL, X. HERES, M. MIGUIRDITCHIAN, I. BISEL, D. ESPINOUX, B. CAMES, C. VIALLESOUBRANNE P. BARON P., B. LORRAIN, "Results of recent counter-current tests on an(iii)/ln(iii) separation using todga extractant", 9th Bi-Annual Scientific World Meeting on the Nuclear Fuel Cycle (NFC) (GLOBAL - 2009), Paris, France, 6 au 11 septembre 2009
[12] M. MIGUIRDITCHIAN, C. SOREL, B. CAMES, I. BISEL, P. BARON, D.ESPINOUX, J.-N. CALOR, C. VIALLESOUBRANNE, B. LORRAIN, M. MASSON, "HA demonstration in the Atalante facility of the Ganex 1st cycle for the selective extraction of Uranium from HLW", Proceedings GLOBAL09, Paris, France, 6 au 11 septembre 2009.
[13] M. MIGUIRDITCHIAN, C. SOREL, B. CAMÈS, I. BISEL, P. BARON, D. ESPINOUX J-N CALOR, C. VIALLESOUBRANNE, B. LORRAIN, M. MASSON, "HA demonstration in the Atalante facility of the Ganex 2nd cycle for the grouped TRU extraction", 9th Bi-Annual Scientific World Meeting on the Nuclear Fuel Cycle (NFC) (GLOBAL - 2009), Paris, France, 6 au 11 septembre 2009.